Promoting circadian rhythms to optimize gut-to-brain signaling to optimize Alzheimer’s disease (Funded R01)

The aims of the projects are to
1) Determine whether the microbial population is different in DrosophilaAD models and if depletion/disruption of gut microbiota negatively impacts AD-associated dysfunction
2)Determine how dysbiosis-induced gut-to-brain signaling occurs in Drosophila models of AD and
3) Timeseries analysis of Drosophila microbiome in AD model under ALF and TRF, and their validation in vivo, including testing the human microbiome and probiotics for treatments.
Time Restricted Feeding (TRF) & Circadian Clocks Mediated Lifestyle Interventions to Mitigate Ageing-linked Cardiometabolic and Muscular Disorders (Funded R01)

The leading risk factors for cardiometabolic diseases are age, shift work, obesogenic challenges, and aberrant eating/sleeping patterns. Each of these factors disrupts circadian rhythms, and perturbation of the circadian clock in model organisms increases the severity of cardiac diseases. Conversely, dietary intervention such as time-restricted feeding (TRF) in which all calories are consumed within a consistent 12-hour or shorter daily interval, without reducing caloric intake, has been shown to prevent various metabolic disorders and attenuate age-related cardiac dysfunction. TRF is known to sustain behavioral and molecular circadian rhythms, but the mechanism by which it improves cardiac health is largely unknown. Therefore, studies in a model like Drosophila that allow specific perturbation of lifestyle and genetic makeup are important for mechanistic studies.
We discovered molecular pathways that preserve age-associated deterioration of cardiac muscle under TRF conditions. Now we are assessing the relevance of circadian rhythm disruptions on cardiac health in Drosophila. We are also evaluating the impact of TRF dietary intervention on the transcriptomics profile in the heart that correlates with improvement in cardiac physiology. Assessing the mechanism underlying the benefits of TRF dietary intervention on cardiac function will provide insight into the primary causes and molecular consequences of pathogenic signaling linked with metabolic and cardiac disorders.
The pleiotropic beneficial effects of TRF in both mammals and flies in mitigating multiple metabolic and cardiac risk factors, without altering nutrition quality or quantity, has opened a potential lifestyle modification strategy to combat cardiometabolic diseases. Future studies will include validation of our Drosophila findings using mouse models. This lifestyle intervention has the potential to treat metabolic syndrome, including cardiometabolic disorders in humans.
Collaborator: Dr. Satchidananda Panda, Salk Institute of Biological Science
Cell-molecular Basis of Cardiomyopathies, Neuropathies, and Aging Linked with Protein Misfolding/Aggregation (Funded MPI R01)

We are using multidisciplinary integrative approaches to investigate the mechanism of amyloid‐induced protein‐unfolding, aggregation, and metabolic defects that lead to cardiomyopathies, diabetes mellitus, muscle atrophy, and inclusion body myopathy in a Drosophila model. Amyloid precursor proteins are prone to misfolding and are involved in cardiac amyloidosis. Amyloidosis constitutes a large group of proteinopathies characterized by the accumulation of misfolded proteins into aggregates in various tissues. Several amyloid‐linked neurodegenerative and metabolic diseases, including Huntington’s disease, Alzheimer’s disease, tauopathy, Parkinson’s disease, amyotrophic lateral sclerosis, and diabetes mellitus, exhibit pathological protein aggregates and are often associated with striated muscle defects. Amyloidosis’ relationship with striated muscle defects and metabolic disorders has been largely overlooked and the mechanisms of amyloid‐related striated muscle and aging disorders are poorly understood.
More recently my research group has been focused on understanding the molecular aspects of the aging process that increase the risk for Alzheimer’s disease (AD) pathology that will enable urgently needed interventions to delay AD onset. AD patients are at a greater risk for multiple diseases and functional decline in peripheral organs, often before the onset of overt neurological disease. Circadian rhythms, which are intimately interlinked with metabolism, orchestrate the coordinated expression and function of multiple pathways that support normal function and repair in both neural and peripheral tissues. Circadian rhythms deteriorate with aging, and Alzheimer’s patients show disrupted circadian rhythm. However, the reciprocal relationship between circadian rhythm disruption (CRD) and AD pathology is unexplored.
To test the molecular pathways mediating the opposite effects of CRD and TRF on AD pathologies, time-series transcriptomes from these flies will be analyzed to find candidate pathways. The functional significance of these pathways will be tested in vivo using genetic modulation. Moreover, we also propose to screen small molecule suppressors to eventually modify potential therapeutic molecules for better efficacy. Successful completion of this project will provide a deeper molecular understanding of the interaction between circadian rhythm and genetic risk of AD. Additionally, this research will assess the efficacy of a behavioral intervention that would have a high potential for adoption in humans.
Collaborators: Dr. Satchidananda Panda, Salk Institute of Biological Science, and Dr. Sanford Bernstein, San Diego State on ultrastructural approach.
Delineating Pathological Pathways Linking Insomnia with Cardiovascular Diseases (Funded MPI R01)

Insomnia disorder, defined by persistent difficulty in initiating or maintaining sleep, and corresponding daytime dysfunction occurs in roughly 10-20% of the population and leads to high socioeconomic costs and substantial lifetime morbidity. Prospective epidemiologic studies suggest that insomnia increases the risk for developing cardiovascular disease (CVD). However, underlying common pathophysiological processes and causal mechanisms remain poorly understood. Importantly, it is unknown whether interventions targeting objective sleep disturbances in insomnia could improve cardiovascular functional outcomes. Recently Dr. Richa Sexana’s lab at the Harvard Medical School has identified and replicated 57 genetic loci for self-reported insomnia symptoms that impact objectively measured short sleep and sleep efficiency in large-scale genome-wide association studies. Shared genetic links were found between insomnia and CVD. Furthermore, Mendelian randomization studies demonstrated a causal link between insomnia symptoms and prevalent and incident CVD across two cohorts (two- to three-fold increased risk of CVD with genetically instrumented case status for insomnia) while no evidence of reverse causality was found.
We propose to define candidate causal genes and variants for insomnia symptoms at 57 genome-wide significant loci and test their impact on CVD risk. We will also test the functional consequences of mutations in orthologous causal insomnia genes on sleep and cardiovascular physiology in Drosophila. We will also test the relationship between sleep disruption and cardiovascular function in Drosophila using genetic, dietary, and pharmacologic perturbations. Using human genetics and fruit fly models, this work will elucidate mechanisms by which insomnia adversely influences CVD and inform on therapeutic strategies both for insomnia and for CVD. Coincident with this, CVD has remained recalcitrant as the leading cause of human mortality.
Collaborators: Drs. Richa Saxena and James Walker (Massachusetts General Hospital and Harvard University)
Mutant-Lamin (LMNA) Based Cardiomyopathies, Skeletal Myopathies, and Lipodystrophies (Pending R01)

Cytoplasmic aggregations of mutant-Lamin in the cardiomyocytes (upper) and myocytes (lower)
Laminopathies are a group of genetic disorders caused by dominant mutations in the human LMNA gene encoding A-type lamins, which form intermediate filaments that line the inner nuclear membrane. Patients with laminopathies exhibit phenotypes of aging, including cardiac and skeletal muscle dysfunction, diabetes, and progeria. Over 400 mutations have been identified in the LMNA gene – the highest number of disease mutations discovered in a single gene. Cardiac failure with aggressive arrhythmias and conduction defects is the major cause of death among individuals with laminopathies. The underlying molecular signalling pathways are not well understood and there are currently no treatments.
We have established Drosophila melanogaster (Drosophila) models to evaluate cardiac and skeletal muscle pathologies caused by mutant lamins. In the fly, synonymous mutations from humans are modeled in the Drosophila Lamin C (LamC) gene, which is analogous to LMNA in humans. Cardiac and skeletal muscle-specific expression of mutant LamC in the fly resulted in compromised cardiac and skeletal muscle functions, mis-regulation of redox homeostasis, and autophagy as well as cytoplasmic aggregation of LamC and other nuclear envelopes (NE) proteins. Moreover, to divulge the relevance of this to human pathology, transcriptomic data obtained from patient muscle biopsies showed alteration of genes linked with redox homeostasis and AMPK pathways. Our genetic validation results confirmed that modulation of redox stress/AMPK suppressed striated muscle laminopathy. To mimic human disease conditions, we are generating novel Drosophila and mouse CRISPR models for mutant LamC.
These studies are innovative because we use integrated approaches to define the mechanistic basis of aggregation-based striated muscle laminopathies. These studies are highly significant as they provide insights into treatments for laminopathy in humans that are linked with aggressive and deadly arrhythmias with conduction defects.
Collaborators: Dr. Lori Wallrath, University of Iowa, and Dr. Rajasekaran Namakkal-Soorappan, UAB, School of Medicine.
Machine-learning approaches for automated quantification of cardiac, skeletal muscle, and sleep/activity data
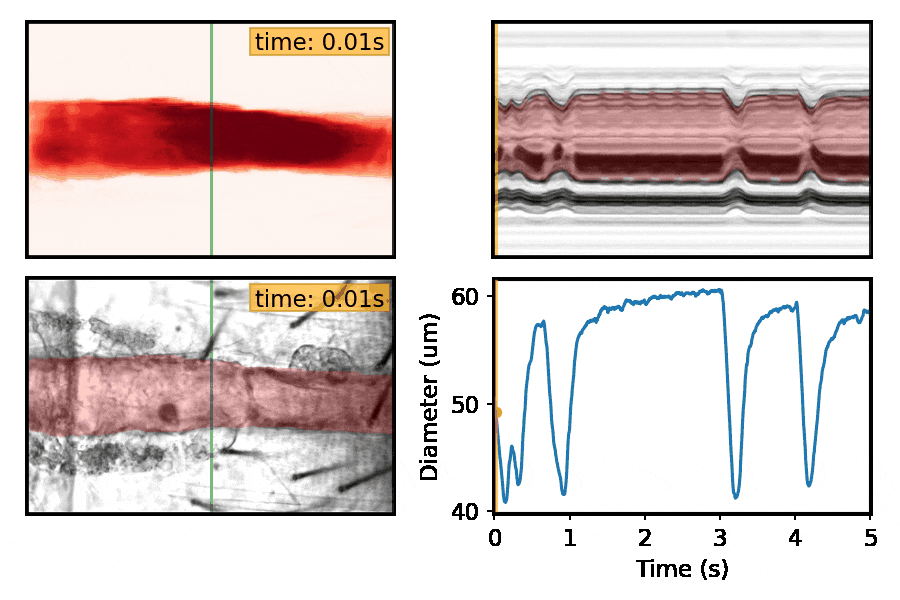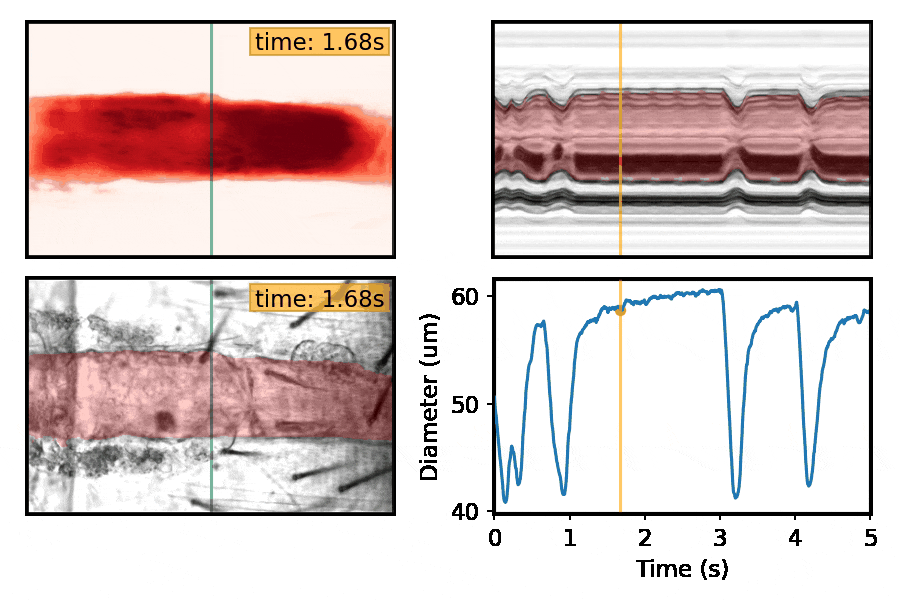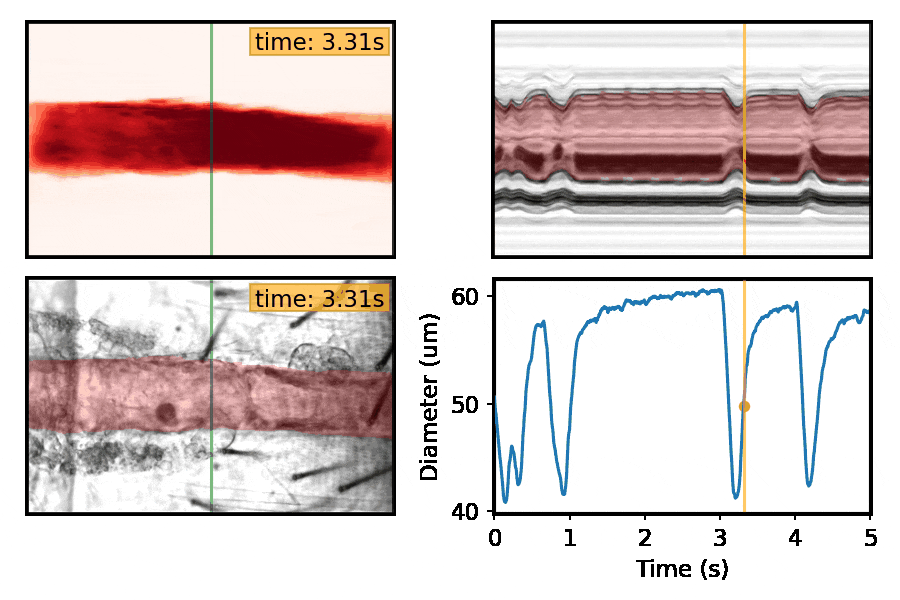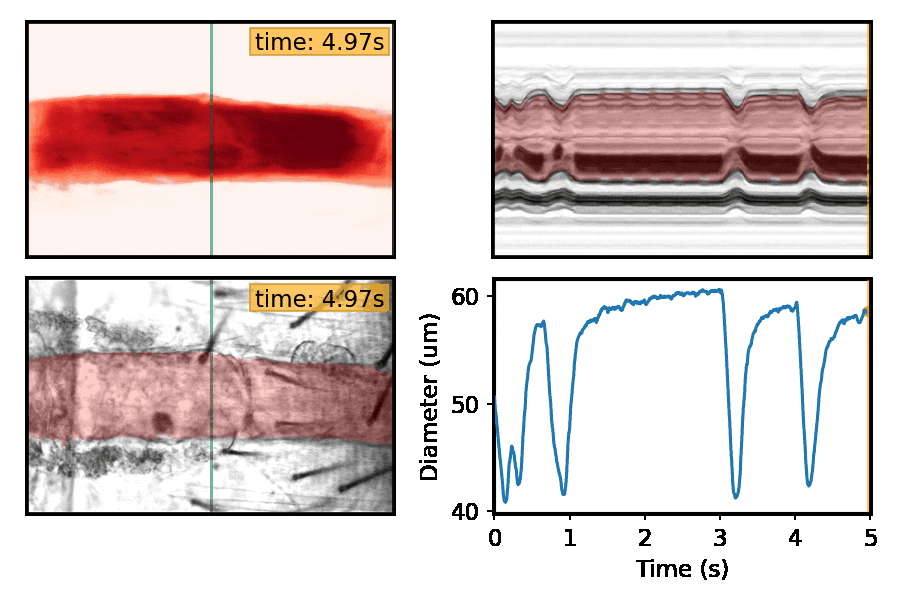
This section is a work in progress. Check back shortly!
Grant Support
Research funding has allowed Dr. Melkani and his personnel to continue researching over the years. Listed below are the current 4 R01 and past grants.
R01 NIH/NIA (2019-2024) “Molecular basis of circadian rhythms disruptions linked cardiometabolic disorders and their mitigation using dietary intervention” (Melkani, PI).
The aims of the projects are to 1) assess the relevance of circadian rhythm organization and TRF mediated intervention on cardiac health in Drosophila, 2) assess the impact of TRF dietary intervention on the temporal gene expression profile in the heart that correlates with improvement in cardiac physiology, and 3) evaluate the mechanism underlying the benefits of TRF dietary intervention on cardiac function.
R01 NIH/NHLBI (2020-2024) “Dissecting the causal role of insomnia in cardiovascular disease” (Melkani, G.C., Saxena, R. and Walker, J. (MPI), Dr. Saxena is contact PI).
The aims of the projects are to 1) define causal genes and pathways for insomnia symptoms at 57 GWAS loci in humans and test their impact on cardiovascular disease, 2) test the consequence of the loss of function of the Drosophila orthologs of causal human insomnia genes on sleep and cardiovascular function, and 3) test the impact of perturbed sleep (disrupted or improved) on cardiovascular function in humans and Drosophila.
R01 NIH/NIA (2020-2024) “Optimized circadian rhythms for the prevention of Alzheimer’s disease” (Melkani, G.C., and Panda, S. (MPI), Dr. Panda is contact PI).
The aims of the projects are to 1) test the reciprocal relationship between Alzheimer’s disease (AD) and circadian rhythm disruption, 2) test the protective effect of Time-Restricted Feeding on age-related AD pathology, and 3) test the molecular mechanism by which TRF attenuates functional decline in AD.
R01 NIH/NIAMS (2013-2018) ”Mechanism of Myosin Chaperone UNC-45: Structural, Functional & Genetic Approaches” (Bernstein, PI; Melkani, Collaborator).
The aims of the project are to 1) investigate the dimerization of unc-45 at a structural level and determine the importance of dimerization in unc-45 function, 2) perform structure-function analysis of a conserved surface cleft of unc-45 that we identified by crystallography and phylogenetic sequence comparisons, and 3) use combined genetic and biochemical approaches to identify other partners and targets of unc-45.
R21 NIH/NIA (2015-2017) “Genetic analysis and regulation of laminopathy induced cardiac defects” (Melkani, PI).
The aims of the projects are to 1) determine the mechanistic basis of cardiac muscle dysfunction associated with different lamin mutants, and 2) identify genetic pathways that modulate laminopathy-associated cardiac defects. Overall, the outcome of the proposed experiments will provide valuable insights into the molecular mechanisms by which mutant lamins cause cardiac defects and provide insights on potential avenues for therapy for premature aging and cardiac defects.
AMC21 2NDR016592 (2022-2023) “Genetic Modulation and Development of Therapeutic Approaches for Cardiac Laminopathy” (Melkani, G.C., PI; Namakkal-Soorappan, Collaborator), Heersink School of Medicine (HSOM), University of Alabama at Birmingham
The aims of the projects are to 1) examine the mechanistic linkages of pathogenic genes/pathways identified from transcriptomic of human muscle biopsies, using genetic modulation that suppresses cardiomyopathy caused by LMNA mutations (R205W and G489V) and 2) determine whether LMNA mutations (i.e. R190W & G449V) induce proteotoxic cardiac disease in novel mouse CRISPR models and assess the impact of Nrf2 diminution/ablation on suppressing cardiomyopathy via AMPK activation.
Collaborator: Dr. Rajasekaran Namakkal-Soorappan, UAB, School of Medicine.
R21 NIH/NIA (2015-2017) “Genetic analysis and regulation of laminopathy induced cardiac defects” (Melkani, PI).
The aims of the projects are to 1) determine the mechanistic basis of cardiac muscle dysfunction associated with different lamin mutants, and 2) identify genetic pathways that modulate laminopathy-associated cardiac defects. Overall, the outcome of the proposed experiments will provide valuable insights into the molecular mechanisms by which mutant lamins cause cardiac defects and provide insights on potential avenues for therapy for premature aging and cardiac defects.
BGIA AHA (2013-2015) “Tau-induced cardiomyopathy in a Drosophila heart model”(Melkani, PI).
The aims of the project are to 1) establish a Drosophila cardiac model to investigate structural and functional defects in the heart caused by hyperphosphorylation of Tau, and 2) identification of genetic and pharmacological suppressors of Tau-mediated cardiomyopathy.
21 NIH/NCRR (2011-2013) ”Genetic analysis and regulation of amyloids flux in the Drosophila heart” (Melkani, PI).
The aims of the project are to 1) express and detect HD- and AD causing amyloids in the Drosophila heart to explore their impact on cardiac physiology, and 2) ameliorate cardiac amyloidosis by overexpression of chaperones and superoxide dismutase (SOD) or by using pharmacological agents.
Postdoctoral Fellowship AHA (2007-2008) “Assessment and Alleviation of Cardiac Structural and Physiological Defects in Drosophila UNC-45 Chaperone Mutants (Melkani, PI).
The aims of the project are to 1) establish the cardiac defects associated with mutation or knockout (RNA interference) of the Drosophila unc-45 gene, and. 2) determine the impact of UNC-45 chaperone’s function during cardiac aging in Drosophila and investigate the effect of chaperone modulators on cardiac performance.
