Project 1.
Pulmonary immunity to viruses, tumors and allergens.
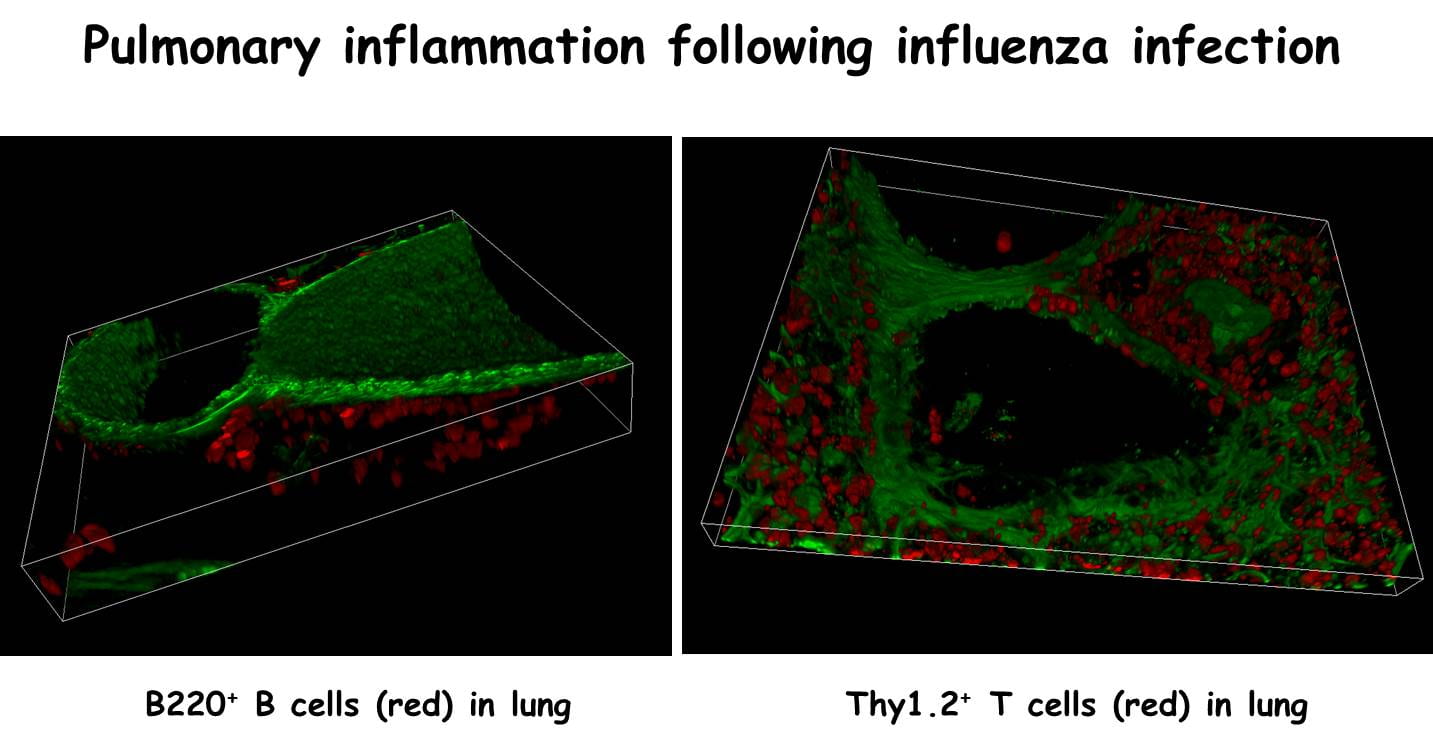
One of the projects in my lab is to determine how local lymphoid tissues develop and function in the respiratory tract. We find that, although naïve mice lack detectable lymphoid areas in their lungs, organized lymphoid tissues are formed in response to infection or inflammation. These tissues, which we have termed “inducible Bronchus Associated Lymphoid Tissue” (iBALT), have separated B and T cell areas, germinal centers, high endothelial venules and specialized dendritic cells. We also find that immune responses to influenza can be generated at sites of iBALT in mice that completely lack lymph nodes, Peyer’s patches and spleen. In fact, mice lacking conventional lymphoid organs are more resistant to influenza than normal mice. Thus, iBALT is not simply an area of inflammation, but is a site that can actively participate in the immune response and appears to regulate respiratory immune responses in a way that leads to less morbidity and mortality. Consistent with this idea, recent data suggests that humans with non-small cell lung carcinoma who also develop iBALT have a better prognosis than those who not. Thus, we are currently exploring ways to induce iBALT in mouse models of lung cancer to determine whether the purposeful induction of iBALT will lead to cancer regression. On the other hand, we also find that iBALT is generated in the lungs of humans with a wide variety of pulmonary diseases. In particular, humans with lung disease associated with rheumatoid arthritis are very prone to developing extensive areas of iBALT. We suspect that these local lymphoid tissues are responding to autoantigens and exacerbating pulmonary disease. Interestingly, we have also found that there is a developmental window in neonatal mice during which iBALT is most easily formed. This window corresponds to a time when children are at risk of developing asthma in response to respiratory viruses, such as RSV. Thus, iBALT is probably involved in both helpful and harmful immune responses in the lung.
Project 2.
Peritoneal immunity to tumors and commensal organisms
A second project in my lab is to understand the development function of unusual lymphoid tissues in the peritoneal and pleural cavities. These tissues include the milky spots of the omentum and the Fat-Associated Lymphoid Clusters (FALC) in the mesentery. The omentum connects the spleen, stomach, pancreas and colon and is a major depot of abdominal fat, which is a source of inflammatory cytokines and chemokines. In addition, the omentum contains milky spots, which collect fluids, particulates and, importantly, metastasizing tumor cells from the peritoneal cavity. We find that milky spots are functional secondary lymphoid organs. However, we also find that the omentum suppresses immune response to peritoneal tumors. Given that a variety of peritoneal tumors, such as ovarian carcinomas and gastrointestinal stromal cell tumors shed cells that metastasize to the omentum, where they seem to grow unchecked due to a combination of profound immunosuppression and exuberant angiogenesis, it is essential that we understand the mechanisms that allow tumor growth in the omentum in order to treat peritoneal tumor metastases appropriately. Our overall hypothesis is that the normal function of the omentum is to maintain tolerance to commensal organisms and food antigens of the gut. Thus, the omentum has evolved to prime CD4 T cells to become Tregs as well as mucosal-type CD4 T cells that produce IL-10 and TGFb and help B cells switch to IgA. However, in the context of tumor antigens, these same functions suppress CD8 T cell responses, promote tolerance to tumor antigens and impair anti-tumor immunity. Thus, my lab is determining the immunological mechanisms by which the omentum suppresses immune response to peritoneal tumors and maintains tolerance to commensal and food antigens.
Project 3.
Control of CD8 T cell responses to viruses and tumors
A third project in my lab is to determine how CD40 signaling controls CD8 T cell responses to viruses and tumors. We find that CD40 signaling is important for both primary and secondary CD8 responses to influenza and tumor antigens. Surprisingly, however, CD4 T cells seem to be dispensable. This scenario is difficult to reconcile with the paradigm that CD40 ligand is expressed by CD4 T cells. Nevertheless, we now have data suggesting that CD4 effector T cells provide CD40 ligand in order to counteract the activities of CD4 Tregs. Thus, in the absence of all CD4 T cells, CD40 ligand is not required. These data suggest that there is a competition between CD4 effectors and Tregs to control the activity of APCs that regulate the priming, expansion and differentiation of CD8 T cells responding to both viruses and tumors. As a first step in testing this hypothesis, we have identified the important APCs (dendritic cell populations) that are required for CD8 T cell responses to influenza and tumor antigens. We are now determining how CD4 effectors and Tregs control the activity/survival/migration of these dendritic cells. Interestingly, different class I restricted epitopes of influenza have different requirements for CD4 cells and CD40 signaling, suggesting that even in a single infection model, each epitope is processed and presented by different cells, which are activated using different pathways. These data will have profound implications on how we vaccinate against specific pathogens and, more importantly, particular epitopes in those pathogens.

Project 4.
Local control of autoimmune/inflamatory T and B cell responses
It is well established that ectopic lymphoid follicles are formed in patients with a variety of autoimmune diseases, including rheumatoid arthiritis, diabetes and multiple sclerosis. These ectopic follicles typically correlate with the most severe forms of disease and are also found at sites of local inflammation or pathology. Thus, most investigators, including us, have concluded that the ectopic follicles are contributing to disease. However, we also find that ectopic follicles promote resistance to infectious disease and reduce inflammation. Therefore, we are trying now to determine the real cause and effect relationship between ectopic follicles and local inflammatory reactions. For example, using mice that have human TNF expressed as a transgene, we showed that these mice develop pulmonary disease, similar to that seen in some RA patients. To test whether the development of ectopic follicles in the lung accelerated disease, we infected the TNF transgenic mice with influenza. Surprisingly, we found that disease was substantially delayed, despite the development of iBALT. Thus, the formation of ectopic follicles may reduce inflammation in autoimmune disease. In another series of experiments, we are using Gaq-/- mice, which spontaneously develop a disease like RA. We are now smoking these mice to determine whether pulmonary exposure to cigarette smoke accelerates disease and/or leads to ectopic follicles in the lung. Finally, we are developing transgenic mice in which we can induce the formation of ectopic follicles in the lungs, brains, pancreas or other organs independently of inflammation. Thus, we can test the ability of ectopic follicles to reduce inflammation in the context of autoimmune disease.
