Current Research
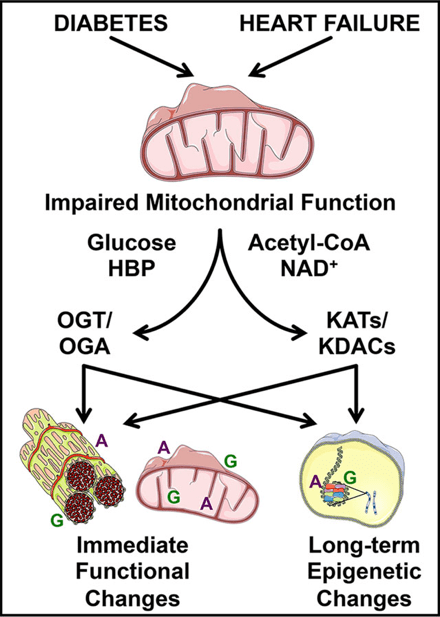
i) PDKs and protein acetylation: Cardiovascular disease is the number one cause of mortality in the United States with different etiologies of heart failure associated with distinct changes in mitochondrial substrate selection and utilization. Here we test the hypothesis that one family of kinases that regulate this process (i.e., PDK) play distinct roles in mortality, cardiac hypertrophy, mitochondrial oxidative metabolism, and signaling to transcriptional pathways in the nucleus mediated by epigenetics (i.e., histone acetylation).
Image from Wende 2016 Proteomics Clin Appl. 10(1):25-38. https://doi.org/10.1002/prca.201500052
We have developed new loss-of-function mouse models of the cardiac isoforms of this pathway (i.e., PDK2 and PDK4) and will utilize specific inhibitors to determine the mechanism of this regulation and test the therapeutic potential of their isoform-specific inhibition in treating pressure-overload induced cardiac hypertrophy and heart failure.
Image from Lopaschuk, Karwi, Tian, Wende, and Abel 2021 Circ Res. 128(10):1487-1513. https://doi.org/10.1161/circresaha.121.318241
Representative Citations
- Dr. Wende’s first area of interest was the regulation of the crosstalk between glucose and fatty acid oxidation with a focus on its regulation via the pyruvate dehydrogenase (PDH) complex by the pyruvate dehydrogenase kinases (PDKs). These two papers and the reviews in the images above lay the foundation for recently funded work (Dr. Ha’s AHA fellowship and Dr. Wende’s latest RO1) expanding upon the crosstalk between glucose/O-GlcNAcylation and now protein acetylation. This project area represents a new area based on a solid foundation.
- Wende AR, Huss JM, Schaeffer PJ, Giguère V and Kelly DP. PGC-1α coactivates PDK4 gene expression via the orphan nuclear receptor ERRα: A mechanism for transcriptional control of muscle glucose metabolism. Molecular and Cellular Biology, 25(24):10684-10694, 2005. iCite NIH% (94.4). PMCID: PMC1316952. 10.1128/MCB.25.24.10684-10694.2005
- Wende AR, Schaeffer PJ, Parker GL, Zechner C, Han DH, Chen MM, Hancock CR, Lehman JJ, Huss JH, McClain DA, Holloszy JO and Kelly DP. A role for the transcriptional coactivator PGC-1α in muscle refueling. The Journal of Biological Chemistry, 282(50):36642-36651, 2007. iCite NIH% (92.3). PMID: 17932032. 10.1074/jbc.M707006200
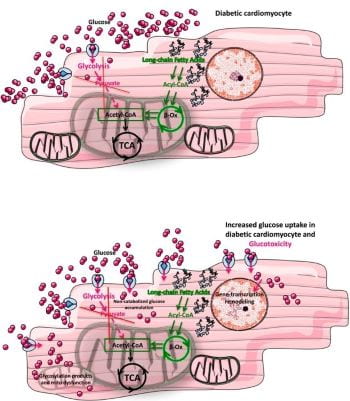
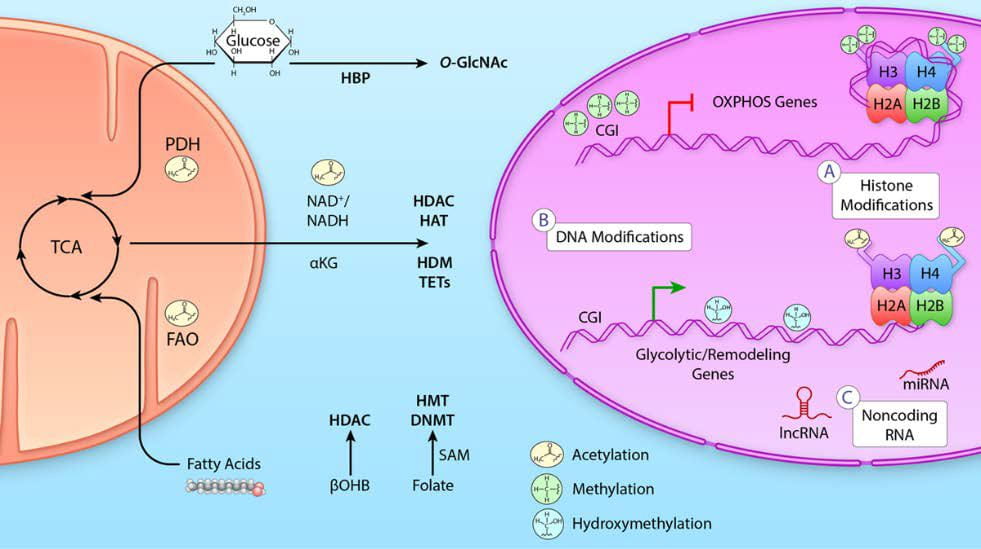
ii) Glucose and GlcNAc: Heart failure is characterized by a decline in mitochondrial oxidative capacity and is a major cause of death in individuals with diabetes. Although several groups have focused on the metabolic changes associated with this owing to decreased glycolytic and oxidative metabolic regulation, we have expanded that to also include cellular signaling. Specifically, it is suggested that glucose flux through the hexosamine biosynthetic pathway leads to altered protein post-translational regulation via O-GlcNAcylation. This process is regulated by two enzymes O-GlcNAc transferase (OGT) to add O-GlcNAc and O-GlcNAcase (OGA) to remove O-GlcNAc from proteins.
Image from commentary on our paper by Gambardella, Lombardi, and Santulli 2020 Diabetes 69:2054-2057. https://doi.org/10.2337/dbi20-0024
To assess both glucose uptake into the cell as well as regulation of proteins directly by enhanced O-GlcNAc levels we have developed two mouse models. Both are inducible for cardiac-specific overexpression of either the glucose transporter 4 (GLUT4; mG4H) or a dominant-negative OGA (dnOGAh). The mG4H model is used to determine the contribution of cellular glucose uptake in the development of mitochondrial dysfunction in the heart, particularly in the context of diabetes. While the dnOGAh model is being used to assess both the role of O-GlcNAc in physiology as well as pathophysiology.
Image from review by Chatham, Zhang, and Wende 2021 Physiol Rev. 101(2):427-293. https://doi.org/10.1152/physrev.00043.2019

Representative Citations
- Earlier work showing that loss-of-function of glucose uptake transitions either physiologic stimuli (i.e., exercise) or pathologic stimuli (i.e., surgically induced pressure-overload) into maladaptive responses.
- Wende AR#, Kim J, Holland WL, Hu P, Wayment BE, Tuinei J, Brahma MK, Pepin ME, McCrory MM, Luptak I, Narra KK, Soto J, Summers SA, Litwin SE and Abel ED#. Glucose transporter 4 (GLUT4) deficient hearts develop maladaptive hypertrophy in response to physiological and pathological stresses. American Journal of Physiology – Heart and Circulatory Physiology, 313(6):H1098-H1108, 2017. PMCID: PMC5814656. 10.1152/ajpheart.00101.2017
- Our study showing that gain-of-function of glucose uptake in the context of hyperglycemia associated with diabetes exacerbates cardiac decline leading to a model of glucotoxicity partly driven by protein O-GlcNAcylation. Also associated commentary for the image above.
- Wende AR#, Schell JC, Ha CM, Pepin ME, Khalimonchuk O, Schwertz H, Pereira RO, Brahma MK, Tuinei J, Contreras-Ferrat A, Wang L, Andrizzi CA, Olsen CD, Bradley WE, Dell’Italia JL, Dillmann WH, Litwin SE and Abel ED#. Maintaining myocardial glucose utilization in diabetic cardiomyopathy accelerates mitochondrial and contractile dysfunction. Diabetes, 69(10):2094-2111, 2020. PMCID: PMC7506832. 10.2337/db19-1057
- Commentary: Gambardella J, Lombardi A and Santulli G. Metabolic flexibility of mitochondria plays a key role in balancing glucose & fatty acid metabolism in the diabetic heart. Diabetes, 69(10):2054-2057, 2020. PMCID: PMC7506829. 10.2337/dbi20-0024
- Extension of our studies showing that either glucose delivery or enhanced protein O-GlcNAcylation replicate some of the molecular changes seen in diabetes (e.g., type 1 or type 2) and reprogram a novel aspect of cardiac metabolism specifically focused on ketone body utilization.
- Brahma MK, Ha CM, Pepin ME, Mia S, Sun Z, Chatham JC, Habegger KM, Abel ED, Paterson ED, Young ME and Wende AR#. Increased glucose availability attenuates myocardial ketone body utilization. Journal of the American Heart Association, 9(15):e013039, 2020. PMCID: PMC7792234. 10.1161/JAHA.119.013039
- Comprehensive review on protein O-GlcNAcylation with close collaborators of our laboratories work. This review covers both the physiological and pathological consequences of this post-translational regulation as well as highlighting several unanswered questions. Dr. Chatham and Dr. Wende have had, and continue to have, several grants and active collaborations together.
- Chatham JC, Zhang J and Wende AR. Role of O-linked N-acetylglucosamine protein modification in cellular (patho)physiology. Physiological Reviews, 101(2):427-493, 2021. PMCID: PMC8428922. 10.1152/physrev.00043.2019

iii) DNA methylation: There is a higher prevalence of cardiovascular disease in patients with history of poor control of glucose levels that in turn alter gene expression through modifications of DNA in the cell. These changes, termed epigenetics, contribute to increased susceptibility to heart failure. To determine if glucose and/or O-GlcNAc contribute to the generation and maintenance of these modifications we have used the two models described above, i.e., mG4H and dnOGAh, to directly determine if the observed changes in DNA methylation, e.g., a specific epigenetic mechanism, increase susceptibility to heart failure, e.g., pressure-overload induced hypertrophy.
Image from the cover of Pepin et al. 2019 Lab Invest. 99(3):371-386. https://doi.org/10.1038/s41374-018-0104-x
We have also started assessing the role of these DNA methylation changes in human heart failure samples to determine which are associated with gene expression changes and may confirm the relevance of this mechanism to the development of heart failure in patients resulting from various known risk factors and disease etiologies.
Image from the cover of Pepin et al.2021 Am J Physiol Heart Circ Physiol. 320(5):H2066-H2079. https://doi.org/10.1152/ajpheart.00036.2021

Representative Citations
- Our first publication on the potential role of defining transcriptional reprogramming in human heart failure using DNA methylation. This study identified a DNA hypermethylation associated with the epigenetic regulator, EZH2, specifically in ischemic heart failure.
- Pepin ME, Ha CM, Crossman DK, Litovsky SH, Varambally S, Barchue JP, Pamboukian SV, Diakos NA, Drakos SG, Pogwizd SM and Wende AR#. Genome-wide DNA methylation changes associated with cardiac transcriptional profiles in human ischemic heart failure. Laboratory Investigation, 99(3):371-386, 2019. PMCID: PMC6515060 Cover Article YouTube Highlight: https://youtu.be/TvoEcF-J-0Y (Starts at ~minute 4:00)
- Our second publication on DNA methylation. This one was a collaborative effort with the laboratory of Dr. Wever-Pinzon. This study also highlights DNA hypermethylation in heart failure, however it focuses on the epigenetic regulator, DNMT1, and compares heart failure to control. Ongoing work between the two groups is defining this further.
- Pepin ME, Drakos S, Ha CM, Tristani-Firouzi M, Selzman CH, Fang JC, Wende AR and Wever-Pinzon O. DNA methylation reprograms cardiac metabolic gene expression in end-stage human heart failure. American Journal of Physiology – Heart and Circulatory Physiology, 317(4):H674-H684, 2019. PMCID: PMC6843013. 10.1152/ajpheart.00016.2019
- Our most recent publication on DNA methylation. This one identified a surprising, but we think important connection to racial disparities, epigenetics, and heart failure. This has also laid the foundation for additional projects focused on etiology specific differences related to race and heart failure susceptibility.
- Pepin ME, Ha CM, Potter LA, Bakshi S, Barchue JP, Haj asaad A, Pogwizd SM, Pamboukian SV, Hidalgo BA, Vickers SM and Wende AR#. Racial and socioeconomic disparity associates with differences in cardiac DNA methylation among men with end-stage heart failure. American Journal of Physiology – Heart and Circulatory Physiology, 320(5):H2066-H2079, 2021. PMCID: PMC8163657 Cover Article. 10.1152/ajpheart.00036.2021
Podcast: https://www.podbean.com/ew/pb-j34vs-103807e
Commentary: Bunsawat K and Robinson AT. Delineating racial and socioeconomic-related health disparities in end-stage heart failure: Insight from cardiac DNA methylation. American Journal of Physiology – Heart and Circulatory Physiology, 320(5):H2031-H2033, 2021. PMCID: PMC8163651. 10.1152/ajpheart.00186.2021
- Pepin ME, Ha CM, Potter LA, Bakshi S, Barchue JP, Haj asaad A, Pogwizd SM, Pamboukian SV, Hidalgo BA, Vickers SM and Wende AR#. Racial and socioeconomic disparity associates with differences in cardiac DNA methylation among men with end-stage heart failure. American Journal of Physiology – Heart and Circulatory Physiology, 320(5):H2066-H2079, 2021. PMCID: PMC8163657 Cover Article. 10.1152/ajpheart.00036.2021
- Brief comment on epigenetics in diabetic cardiomyopathy.
- Pepin ME and Wende AR#. Epigenetics in the development of diabetic cardiomyopathy. Epigenomics, 11(5):469-472, 2019. PMID: 30895816. 10.2217/epi-2019-0027
- Pepin ME and Wende AR#. Epigenetics in the development of diabetic cardiomyopathy. Epigenomics, 11(5):469-472, 2019. PMID: 30895816. 10.2217/epi-2019-0027
