Investigating Translation Termination
Translation occurs in three main steps. First, translation is initiated when the ribosome assembles onto an mRNA at the translational start site. This is followed by the elongation step where tRNAs decode each codon and each encoded amino acid is sequentially linked together to form a protein. Finally, the termination step is signaled when the ribosome encounters a termination signal in the mRNA, prompting release of the newly made protein.
My research focuses mainly on the termination step of translation. This is the least characterized stage of translation and recent studies have shown this step is more complex than originally thought. We are exploring the factors involved in mediating termination, the mechanisms associated with terminating translation, and how translation termination regulates other cellular processes such as mRNA stability, ribosomal recycling, and translation efficiency. All of these processes are important in regulating gene expression and may also be considered therapeutic targets for a variety of different diseases, including rare genetic diseases, cancer, and infectious diseases.
Suppressing Termination as a Therapeutic Strategy
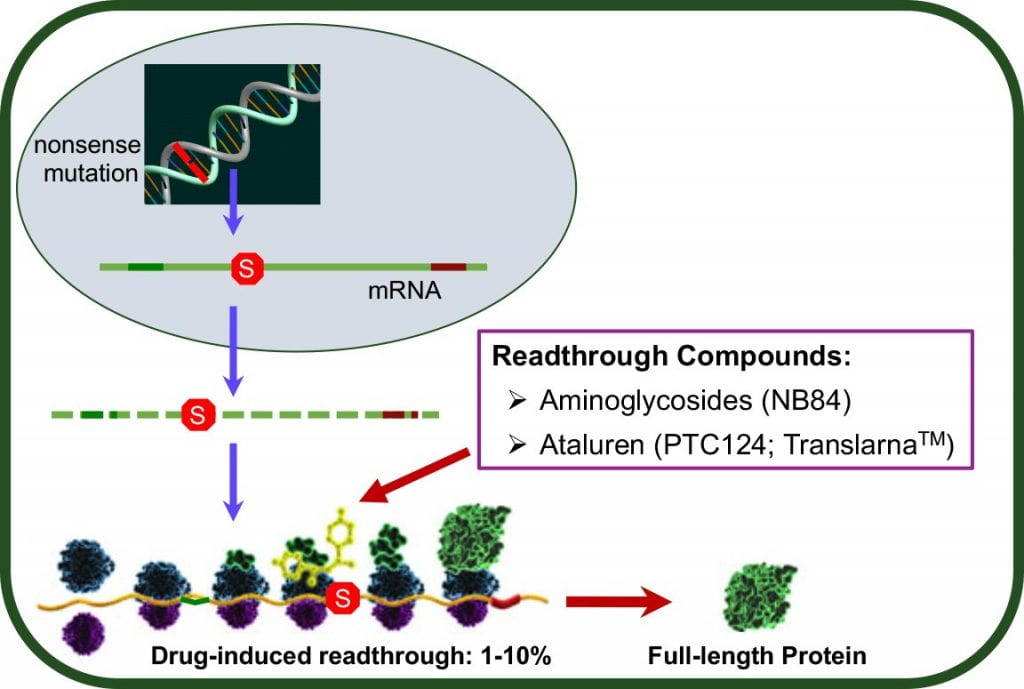
We are exploring translation termination as a therapeutic target to develop treatments for genetic diseases resulting from nonsense mutations. Nonsense mutations are a class of mutation that introduces a premature termination signal into an mRNA, preventing a full-length, functional protein from being translated. We hypothesize that suppressing translation termination at a premature termination signal can restore the production of full-length, functional protein. Our aim is to restore enough functional protein to alleviate genetic diseases caused by nonsense mutations.
We are currently performing high throughput screens of small molecules to identify compounds that suppress termination at premature termination codons (PTCs). We are exploring whether hits from these screens can suppress PTCs and restore deficient protein in a variety of disease models.
Current Diseases We Are Studying
Cystic Fibrosis
Cystic fibrosis (CF) is an autosomal recessive disorder caused by mutations in the Cystic Fibrosis Transmembrane Regulator (CFTR) gene, which codes for a protein that functions as a chloride channel in epithelial cells. Without CFTR function, chloride transport is dysregulated, leading to a build-up of mucus, increased frequency of infections, inflammation, respiratory failure, and other complications. While new modulator compounds are now available to treat defects in CFTR function associated with many different types of mutations, including the prevalent delF508, no treatments are currently available for patients who carry PTC-forming mutations. Significantly, 10% of CF patients carry a nonsense mutation. Click here for more information.
Mucopolysaccharidosis I-Hurler
Mucopolysaccharidosis I (MPS I) is an autosomal recessive lysosomal storage disease caused by a deficiency of alpha-L-iduronidase, an enzyme encoded by the IDUA gene. Severe depletion of alpha-L-iduronidase activity leads to MPS I-Hurler or Hurler syndrome, the most severe form of MPS I. Alpha-L-iduronidase helps break down long chains of sulfated polysaccharides called glycosaminoglycans (GAGs). Without alpha-L-iduronidase, the GAGs dermatan sulfate and heparan sulfate progressively accumulate in lysosomes, eventually leading to the onset of multiple biochemical, morphological, and functional defects in spleen, liver, heart, bone, and neurological tissues. Current treatments for MPS I-H, including stem cell transplantation and enzyme replacement therapy, cannot adequately access the brain, bone, eye, and heart valves and thus, cannot alleviate MPS I-H phenotypes in these tissues. 70% of MPS I-H patients carry a nonsense mutation. Click here for more information.
Mucolipidosis II
Mucolipidosis II, also known as I-cell disease, is a caused by mutations in the GNPTAB gene that codes for the enzyme, UDP-N-acetyleglucosamine-1-phosphotransferase, which attaches GlcNAc-1-P to mannose residues of multiple lysosomal enzymes, allowing them to be targeted to lysosomes. Without GNPTAB activity, multiple lysosomal enzymes do not enter the lysosome, causing their substrates, glycosaminoglycans and sphingolipids, to accumulate in lysosomes. The progressive accumulation of these enzyme substrates leads to phenotypes that are similar to MPS I-H, includes abnormalities in the bone, joints, heart, and neurological tissues. Click here for more information.
Rett Syndrome
Rett syndrome (RTT) is an X-linked neurodegenerative disorder that mainly affects females. RTT is caused by mutations in MECP2, which encodes a protein that binds to methylated chromatin to activate or repress transcription of genes required for neuron maturation. Click here for more information.
FOXG1 Syndrome
FOXG1 Syndrome is caused by mutations in the FOXG1 gene, which encodes the forkhead box G1 protein, a transcription factor that controls cerebrum development. The loss of FOXG1 results in a neurodegenerative disorder with phenotypes similar to those observed with Rett syndrome. However, FOXG1 occurs in both males and females. Click here for more information.
CDKL5 Deficiency Disorder
CDKL5 Deficiency Disorder is an X-linked disorder that results from mutations in the CDKL5 gene that encodes the cyclin-dependent kinase-like 5 protein. CDKL5 protein phosphorylates factors that are essential for normal brain development. Loss of CDKL5 function results in a neurodegenerative disorder with phenotypes that resemble those of Rett syndrome. Click here for more information.
Neurofibromatosis I
Neurofibromatosis I (NF1) is a genetic disorder caused by mutations in the NF1 gene, which codes for neurofibromin. Neurofibromin is expressed in nerve cells, where it acts as a tumor suppressor. Loss of neurofibromin leads to dysregulation of normal cell growth and division, resulting in the formation of tumors called neurofibromas along nerves throughout the body and to the onset of skeletal abnormalities. NF1 patients also have an increased risk of developing other cancers. Click here for more information.


