Research Projects
The Physiology Based Project: Lysine Acetylation and the Regulation of Vasopressin/Aquaporin System in the Principal Cell
Life on land requires that we consume lots of water so that we stay in fluid-balance. If you go on a hike on a warm day, you’ve probably noticed that you sweat, breath heavier and produce very little urine. Our bodies are very good at letting us know when we need to consume and conserve water. If hiking and you don’t drink any water, your plasma osmolality will rise and this is sensed by our nerves and brain. This rise in plasma osmolality will cause the hypothalamus to produce the hormone, arginine vasopressin, and it is secreted from the pituitary gland. Vasopressin quickly travels to our kidneys where it acts on the basolateral membrane of the collecting duct principal cell and ultimately leads to increased water channels (Aquaporins) in these cells that will reabsorb more water. This will then help dilute the plasma and osmolality will return to a normal level.

Aquaporins form small pores in the membranes of our cells that allow the diffusion of water (and sometimes other molecules). In the collecting duct principal cell there is AQP2 on the apical side of the cell and AQP3 and AQP4 on the basolateral side.
These water channels work together to bring water from the urine back into the body. AQP2 and AQP3 are regulated by vasopressin. There is a lot of interesting information on AQP2 and how post-translational phosphorylation of it regulates its expression in the apical membrane (read more here). However, comparatively we know less about the regulation of AQP3.
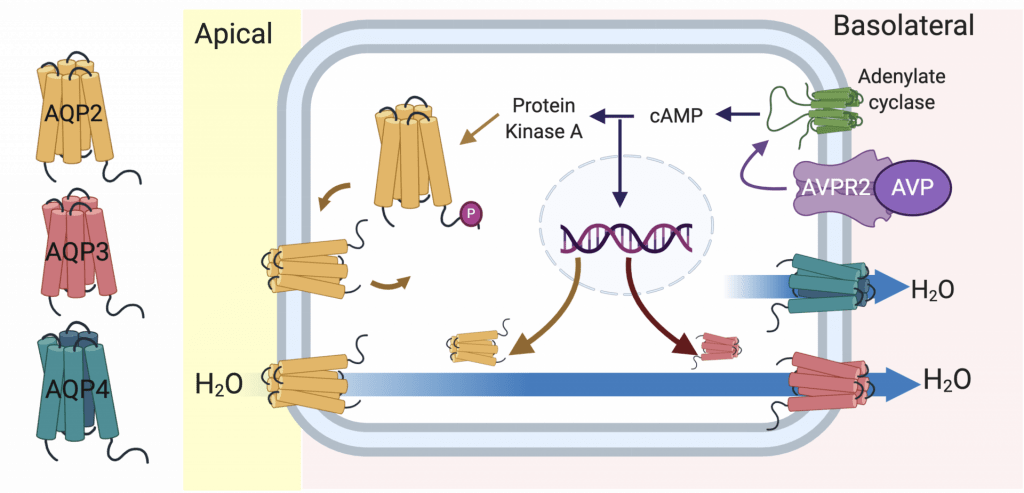
Our lab recently reported the first confirmed post-translational modification of AQP3 acetylation of lysine 282 (read about it here). We are currently determining if this modification affects basolateral AQP3 expression and water permeability.

Immunogold labelling of AQP3 and visualized with electron microscopy. In the basolateral side there are small, black, dots that represent gold particles bound to AQP3. Notice in the apical membrane we don’t observe AQP3, thus normally this water channel is in the basolateral membrane.
The Kidney Disease Focused Project: Epigenetic Regulation of Kidney Fibrosis following AKI
Epigenetics is the study of “on top of” our genetics. It involves various mechanisms that lead to changes in which genes are expressed in our cells. Our DNA is wrapped around proteins called histones, and histones have many lysines that undergo post-translational acetylation/deacetylation. Modifying these histones results in the DNA unwinding and allowing transcription to occur or vice versa, it wraps tightly to the histone and transcription will stop.

There are many epigenetic regulators with high expression in our kidneys (read about them here) and our lab is currently focused on the histone deacetylases (HDACs). Excessive HDAC activity causes cancer and there are currently a few FDA approved HDAC inhibitors used to treat T cell lymphomas and myelomas. In kidney diseases, such as acute kidney disease (short term loss of kidney function) and chronic kidney disease (permanent decline in kidney function), there may be activation of HDACs in our kidneys. But is all HDAC activity bad? Our lab is trying to answer that question. Interstitial fibrosis, which is scarring in our kidneys, is an issue that overtime if it continues to develop can lead to chronic kidney disease. We have identified that HDAC1 is expressed in the kidney and that HDAC1 activation following acute kidney injury can lead to interstitial fibrosis.
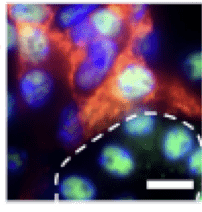
Myofibroblasts are cells that lay down extracellular matrix and can lead to excessive interstitial fibrosis. We can identify them in the kidney by localization of the protein PDGFR beta (in red). These cells also express HDAC1 (green). Outlined in white is a tubule and the tubular cells are also positive for HDAC1 (green). HDAC1 is predominantly in the nucleus.
Using a variety of techniques from cells to whole animal models, we are determining the potential mechanisms involved in HDAC1-mediated interstitial fibrosis!
